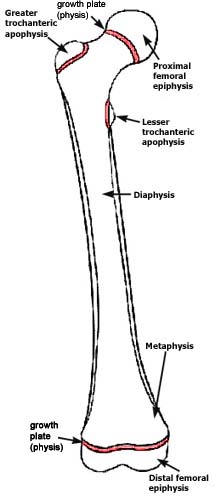
Background and Goals of the
Wilcox lab:
The increase in height in animals is caused by a process called
endochondral ossification. Cartilage
near the end of the bones increases in length and is replaced by bone. This process is very complicated, not
completely understood, and regulated by many genes and growth factor
systems. Hereditary variations in the
different regulatory systems largely determine how tall you will be. When growth ceases after puberty, the growth
cartilage disappears.
Achondroplasia, the related milder disorder hypochondroplasia, and the
lethal disorder thanatophoric dysplasia are caused by mutations in the gene for
Fibroblast growth factor receptor 3 (FGFR3).
All the mutations turn on the receptor excessively. The more the receptor is turned on, the more
severe the dwarfism. Although there
seems to be no excess cancer risk with the mild achondroplasia and
hypochondroplasia mutations, the mutations in the lethal thanatophoric
dysplasia have been associated with several cancers. Why does excessive activity of FGFR3 cause
more growth in some tissues but it causes growth cartilage to slow down how
fast it increases in length? The answer
is still not clear.
FGFR3 is one of the major brakes on growth in cartilage. How FGFR3 functions in cartilage cells is
being sorted out in our lab, Dr. Horton’s lab, and a few others. FGFR3 uses many pathways to cause its effects
in cartilage cells and interacts with a number of the other important signaling
pathways. Some of the pathways we are
just discovering now.
C-type natriuretic peptide (CNP) has been investigated by Dr. Kazuwa
Nakao’s group in Kyoto, Japan. They
found that increasing CNP in the mouse could partially correct dwarfism in the
achondroplasia mouse model. We went on
to elucidate some of the mechanisms for CNP action in cartilage cells and how
it could partially block FGFR3’s actions.
Dr. Krejci and I took the CNP idea to biotechnology companies and
interested BioMarin in the project. They
have gone on to develop a more stable form of CNP that has been shown to
improve growth in a mouse model for achondroplasia and increase growth in normal
monkeys.
While CNP is a significant advance, it can only affect half of what
FGFR3 is doing in cartilage cells and daily injections for the entire period a
child is growing are inconvenient. A longer
lasting treatment or an oral drug would be ideal. The challenge is to identify such a drug that
will not cause significant side effects.
Dr. Krejci developed a drug screening method to identify drugs that
interfere with FGFR3 and are not toxic to cells. Using this method in a small scale screen of
5000 compounds, he identified NF449. It
and its relatives inhibit FGFR3 very well.
However, the structure of the NF449 and its relatives are such that it
won’t make a good oral drug- it doesn’t get into cells easily by itself. A larger drug screen (90,000+ compounds)
should allow us to find better compounds.
Clinical Studies:
People with achondroplasia tend to be obese and have high blood
pressure, which is usually not properly treated. These factors alone may be enough to explain
the fact that achondroplasts tend to die 5-10 years younger. However, FGFR3 is important in many areas of
the body besides the growing cartilage.
We are currently conducting a study on adults with achondroplasia to
find out if they are prediabetic and/or have abnormalities of their blood
vessels.
This drug related to CNP has been tested for
safety in adults. Trials in children
with achondroplasia are expected to start later in 2013. The drug will
have to be injected every day,
much like growth hormone (which is ineffective for increasing growth in
dwarfs). Dr. Wilcox will be one of the
investigators in the trials. Interested families with achondroplastic
children should contact a local site for more information. The
precursor study is a measurement study. Some patients in this study
will be eligible to enter the drug trial. The link for the measurement
study is:
http://www.clinicaltrials.gov/ct2/show/NCT01603095?term=NCT01603095&rank=1
For more information on these clinical studies, contact Tara Funari,
M.S. at (310) 423-9915, email:
tara.funari@cshs.org
Meet the lab:
Pavel Krejci, Ph.D.

Dr. Krejci was born in what is now the Czech Republic. He graduated with a bachelor’s degree in
Biology from the Masaryk University in Brno.
He then went on to obtain his Ph.D. in Molecular Embryology at the Mendel
University in Brno. His graduate work
concentrated on the role of fibroblast growth factors in leukemia. He did post-doctoral fellowships in the
Wilcox laboratory from 2001-2003 and in Toulouse, France from 2003-2004. He then returned to Cedars-Sinai where he is
a research scientist and member of the genetics faculty. He is an Adjunct Associate Professor of
Pediatrics through the UCLA School of Medicine.
Dr. Krejci has maintained his connections in the Czech Republic, where
he is a research scientist and supervises graduate students in the Institute of
Experimental Biology at Masaryk University in Brno. He has obtained several grants in the Czech
Republic to study FGF signaling and, now that he is a permanent resident of the
U.S., applied for grant funding from the NIH.
The ongoing relationship between the U.S. and Czech Republic labs has
been extremely helpful to our ongoing efforts to understand FGFR3 and find
novel targets for treatment.
Dr. Krejci will continue to dissect the pathways FGFR3 uses to cause
dwarfism so new targets can be identified.
Pavel has two children and enjoys hiking, backpacking, fishing, and is
an accomplished naturalist.
Yuan Xue, M.D., Ph.D.
Dr. Xue was born in Wuhan, China.
In 2000, she obtained her M.D. degree from the Tongji Medical University
in Wuhan. She then came to the U.S.
where she obtained her Ph.D. in 2006 from Kansas State University in Human
Nutrition in Molecular Biochemistry.
After her Ph.D., she worked in two core laboratories at UCLA and
Cedars-Sinai from 2007 to 2011.
In 2011, Dr. Xue joined the laboratory to study a treatment for
achondroplasia being tested by a biotechnology company.
Dr. Xue will be conducting a large scale drug screen using a
modification of the screening system that Dr. Krejci developed and used to
identify NF449. The goal is to identify
potential new drugs to treat achondroplasia.
Yuan enjoys hiking, traveling, art, and painting.
Jorge Martin, B.S.
Jorge joined the lab in 2011 as a senior technician after working for
many years at U.C. Irvine. Jorge is in
charge of the tissue and cell collection from the International Skeletal
Dysplasia Registry at Cedars-Sinai, an invaluable resource for studying
dwarfisms.
Jorge will be assisting Drs. Krejci and Xue with some of their work in
the lab.
Jorge is an accomplished musician.
Publications from the
Wilcox/Krejci Laboratory
Fibroblast growth factors:
Krejci P, Pejchalova K, Rosenbloom BE, Rosenfelt FP, Tran EL, Laurell
H,
Wilcox WR. The antiapoptotic protein Api5 and its partner, high
molecular weight
FGF2, are up-regulated in B cell chronic lymphoid leukemia. J Leukoc
Biol. 2007
Dec;82(6):1363-4. Epub 2007 Sep 7. PubMed PMID: 17827341.
Krejci P, Krakow D, Mekikian PB, Wilcox WR. Fibroblast growth factors
1, 2,
17, and 19 are the predominant FGF ligands expressed in human fetal
growth plate
cartilage. Pediatr Res. 2007 Mar;61(3):267-72. PubMed PMID: 17314681.
Krejci P, Mekikian PB, Wilcox WR. The fibroblast growth factors in
multiple
myeloma. Leukemia. 2006 Jun;20(6):1165-8. PubMed PMID: 16598309.
Fibroblast growth factor
signaling:
Krejci P, Aklian A, Kaucka M, Sevcikova E, Prochazkova J, Masek JK, Mikolka P,
Pospisilova T, Spoustova T, Weis M, Paznekas WA, Wolf JH, Gutkind JS, Wilcox WR,
Kozubik A, Jabs EW, Bryja V, Salazar L, Vesela I, Balek L. Receptor Tyrosine Kinases Activate Canonical WNT/β-Catenin Signaling via MAP Kinase/LRP6 Pathway and Direct β-Catenin Phosphorylation. PLoS One. 2012;7(4):e35826. PMCID: PMC3338780 PMID: 22558232
Merrill AE, Sarukhanov A, Krejci P, Idoni B, Camacho N, Estrada KD,
Lyons KM,
Deixler H, Robinson H, Chitayat D, Curry CJ, Lachman RS, Wilcox WR,
Krakow D.
Bent Bone Dysplasia-FGFR2 type, a Distinct Skeletal Disorder, Has
Deficient
Canonical FGF Signaling. Am J Hum Genet. 3012 Mar 9;90(3):550-557. Epub
2012 Mar
1. PubMed PMID: 22387015.
Červenka I, Wolf J, Mašek J, Krejci P, Wilcox WR, Kozubík A, Schulte G,
Gutkind JS, Bryja V. Mitogen-activated protein kinases promote
WNT/beta-catenin
signaling via phosphorylation of LRP6. Mol Cell Biol. 2011
Jan;31(1):179-89. Epub
2010 Oct 25. PubMed PMID: 20974802; PubMed Central PMCID: PMC3019858.
Krejci P, Prochazkova J, Smutny J, Chlebova K, Lin P, Aklian A, Bryja
V,
Kozubik A, Wilcox WR. FGFR3 signaling induces a reversible senescence
phenotype
in chondrocytes similar to oncogene-induced premature senescence. Bone.
2010
Jul;47(1):102-10. Epub 2010 Mar 31. PubMed PMID: 20362703; PubMed
Central PMCID:
PMC3087869.
Salazar L, Kashiwada T, Krejci P, Muchowski P, Donoghue D, Wilcox WR,
Thompson
LM. A novel interaction between fibroblast growth factor receptor 3 and
the p85
subunit of phosphoinositide 3-kinase: activation-dependent regulation
of ERK by
p85 in multiple myeloma cells. Hum Mol Genet. 2009 Jun
1;18(11):1951-61. Epub
2009 Mar 13. PubMed PMID: 19286672; PubMed Central PMCID: PMC2902846.
Krejci P, Salazar L, Kashiwada TA, Chlebova K, Salasova A, Thompson LM,
Bryja
V, Kozubik A, Wilcox WR. Analysis of STAT1 activation by six FGFR3
mutants
associated with skeletal dysplasia undermines dominant role of STAT1 in
FGFR3
signaling in cartilage. PLoS One. 2008;3(12):e3961. Epub 2008 Dec 17.
PubMed
PMID: 19088846; PubMed Central PMCID: PMC2597732.
Krejci P, Prochazkova J, Bryja V, Jelinkova P, Pejchalova K, Kozubik A,
Thompson LM, Wilcox WR. Fibroblast growth factor inhibits interferon
gamma-STAT1
and interleukin 6-STAT3 signaling in chondrocytes. Cell Signal. 2009
Jan;21(1):151-60. Epub 2008 Oct 12. PubMed PMID: 18950705; PubMed
Central PMCID:
PMC2655766.
Matsushita T, Wilcox WR, Chan YY, Kawanami A, Bükülmez H, Balmes G,
Krejci P,
Mekikian PB, Otani K, Yamaura I, Warman ML, Givol D, Murakami S. FGFR3
promotes
synchondrosis closure and fusion of ossification centers through the
MAPK
pathway. Hum Mol Genet. 2009 Jan 15;18(2):227-40. Epub 2008 Oct 15.
PubMed PMID:
18923003; PubMed Central PMCID: PMC2638772.
Krejci P, Salazar L, Goodridge HS, Kashiwada TA, Schibler MJ, Jelinkova
P,
Thompson LM, Wilcox WR. STAT1 and STAT3 do not participate in
FGF-mediated growth
arrest in chondrocytes. J Cell Sci. 2008 Feb 1;121(Pt 3):272-81. Epub
2008 Jan
15. PubMed PMID: 18198189.
Krejci P, Masri B, Salazar L, Farrington-Rock C, Prats H, Thompson LM,
Wilcox
WR. Bisindolylmaleimide I suppresses fibroblast growth factor-mediated
activation
of Erk MAP kinase in chondrocytes by preventing Shp2 association with
the Frs2
and Gab1 adaptor proteins. J Biol Chem. 2007 Feb 2;282(5):2929-36. Epub
2006 Dec
4. PubMed PMID: 17145761.
Drugs and drug screening
technique:
Scuto A, Krejci P, Popplewell L, Wu J, Wang Y, Kujawski M, Kowolik C,
Xin H,
Chen L, Wang Y, Kretzner L, Yu H, Wilcox WR, Yen Y, Forman S, Jove R.
The novel
JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression
of human myeloma cell growth and survival. Leukemia. 2011
Mar;25(3):538-50. Epub
2010 Dec 17. PubMed PMID: 21164517; PubMed Central PMCID: PMC3216671.
Krejci P, Murakami S, Prochazkova J, Trantirek L, Chlebova K, Ouyang Z,
Aklian
A, Smutny J, Bryja V, Kozubik A, Wilcox WR. NF449 is a novel inhibitor
of
fibroblast growth factor receptor 3 (FGFR3) signaling active in
chondrocytes and
multiple myeloma cells. J Biol Chem. 2010 Jul 2;285(27):20644-53. Epub
2010 May
3. PubMed PMID: 20439987; PubMed Central PMCID: PMC2898326.
Krejci P, Pejchalova K, Wilcox WR. Simple, mammalian cell-based assay
for
identification of inhibitors of the Erk MAP kinase pathway. Invest New
Drugs.
2007 Aug;25(4):391-5. Epub 2007 Apr 26. PubMed PMID: 17458503.
Krejci P, Masri B, Fontaine V, Mekikian PB, Weis M, Prats H, Wilcox WR.
Interaction of fibroblast growth factor and C-natriuretic peptide
signaling in
regulation of chondrocyte proliferation and extracellular matrix
homeostasis. J
Cell Sci. 2005 Nov 1;118(Pt 21):5089-100. Epub 2005 Oct 18. PubMed
PMID:
16234329.
Krejci P, Bryja V, Pachernik J, Hampl A, Pogue R, Mekikian P, Wilcox
WR. FGF2
inhibits proliferation and alters the cartilage-like phenotype of RCS
cells. Exp
Cell Res. 2004 Jul 1;297(1):152-64. PubMed PMID: 15194433.
Clinical and molecular:
Danielpour M, Wilcox WR, Alanay Y, Pressman BD, Rimoin DL. Dynamic
cervicomedullary cord compression and alterations in cerebrospinal
fluid dynamics
in children with achondroplasia. Report of four cases. J Neurosurg. 2007
Dec;107(6 Suppl):504-7. PubMed PMID: 18154022.
Schweitzer DN, Graham JM Jr, Lachman RS, Jabs EW, Okajima K, Przylepa
KA,
Shanske A, Chen K, Neidich JA, Wilcox WR. Subtle radiographic findings
of
achondroplasia in patients with Crouzon syndrome with acanthosis
nigricans due to
an Ala391Glu substitution in FGFR3. Am J Med Genet. 2001 Jan
1;98(1):75-91.
Review. PubMed PMID: 11426459.
Kitoh H, Brodie SG, Kupke KG, Lachman RS, Wilcox WR. Lys650Met
substitution
in the tyrosine kinase domain of the fibroblast growth factor receptor
gene
causes thanatophoric dysplasia Type I. Mutations in brief no. 199.
Online. Hum
Mutat. 1998;12(5):362-3. PubMed PMID: 10671061.
Bellus GA, Bamshad MJ, Przylepa KA, Dorst J, Lee RR, Hurko O, Jabs EW,
Curry
CJ, Wilcox WR, Lachman RS, Rimoin DL, Francomano CA. Severe
achondroplasia with
developmental delay and acanthosis nigricans (SADDAN): phenotypic
analysis of a
new skeletal dysplasia caused by a Lys650Met mutation in fibroblast
growth factor
receptor 3. Am J Med Genet. 1999 Jul 2;85(1):53-65. PubMed PMID:
10377013.
Brodie SG, Kitoh H, Lachman RS, Nolasco LM, Mekikian PB, Wilcox WR.
Platyspondylic lethal skeletal dysplasia, San Diego type, is caused by
FGFR3
mutations. Am J Med Genet. 1999 Jun 11;84(5):476-80. PubMed PMID:
10360402.
Tavormina PL, Bellus GA, Webster MK, Bamshad MJ, Fraley AE, McIntosh I,
Szabo
J, Jiang W, Jabs EW, Wilcox WR, Wasmuth JJ, Donoghue DJ, Thompson LM,
Francomano
CA. A novel skeletal dysplasia with developmental delay and acanthosis
nigricans
is caused by a Lys650Met mutation in the fibroblast growth factor
receptor 3
gene. Am J Hum Genet. 1999 Mar;64(3):722-31. PubMed PMID: 10053006;
PubMed
Central PMCID: PMC1377789.
Brodie SG, Kitoh H, Lipson M, Sifry-Platt M, Wilcox WR. Thanatophoric
dysplasia type I with syndactyly. Am J Med Genet. 1998 Nov
16;80(3):260-2. PubMed
PMID: 9843049.
Kitoh H, Lachman RS, Brodie SG, Mekikian PB, Rimoin DL, Wilcox WR.
Extra
pelvic ossification centers in thanatophoric dysplasia and
platyspondylic lethal
skeletal dysplasia-San Diego type. Pediatr Radiol. 1998
Oct;28(10):759-63. PubMed
PMID: 9799297.
Wilcox WR, Tavormina PL, Krakow D, Kitoh H, Lachman RS, Wasmuth JJ,
Thompson
LM, Rimoin DL. Molecular, radiologic, and histopathologic correlations
in
thanatophoric dysplasia. Am J Med Genet. 1998 Jul 7;78(3):274-81.
PubMed PMID:
9677066.
Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS,
Wilcox
WR, Rimoin DL, Cohn DH, Wasmuth JJ. Thanatophoric dysplasia (types I
and II)
caused by distinct mutations in fibroblast growth factor receptor 3.
Nat Genet.
1995 Mar;9(3):321-8. PubMed PMID: 7773297.
Reviews:
Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen years and
counting: the
current understanding of fibroblast growth factor receptor 3 (FGFR3)
signaling in
skeletal dysplasias. Hum Mutat. 2012 Jan;33(1):29-41. doi:
10.1002/humu.21636.
Epub 2011 Nov 16. PubMed PMID: 22045636; PubMed Central PMCID:
PMC3240715.
Krejci P, Prochazkova J, Bryja V, Kozubik A, Wilcox WR. Molecular
pathology of
the fibroblast growth factor family. Hum Mutat. 2009 Sep;30(9):1245-55.
Review.
PubMed PMID: 19621416; PubMed Central PMCID: PMC2793272.
Chlebova K, Bryja V, Dvorak P, Kozubik A, Wilcox WR, Krejci P. High
molecular
weight FGF2: the biology of a nuclear growth factor. Cell Mol Life Sci.
2009
Jan;66(2):225-35. Review. PubMed PMID: 18850066; PubMed Central PMCID:
PMC3229932.
Pejchalova K, Krejci P, Wilcox WR. C-natriuretic peptide: an important
regulator of cartilage. Mol Genet Metab. 2007 Nov;92(3):210-5. Epub
2007 Aug 6.
Review. PubMed PMID: 17681481.
Passos-Bueno MR, Wilcox WR, Jabs EW, Sertié AL, Alonso LG, Kitoh H.
Clinical
spectrum of fibroblast growth factor receptor mutations. Hum Mutat.
1999;14(2):115-25. Erratum in: Hum Mutat 2001 May;17(5):431. PubMed
PMID:
10425034.